Thanks for sharing this, it was a very interesting read!
I do want to question this claim though:
"But the alternative to a project like the AMA is that essential HIV treatments arrive half a decade late in places that needed them most."
It seems to me that an attractive alternative would be for African countries to simply give up on doing their own drug approvals, and outsource the decision making entirely to the FDA, MHRA, PMDA, EMA etc. Why not simply say that any drug approved by any of these agencies is automatically approved? This way drugs would be approved swiftly and with almost zero cost to both government and corporation.
100% agree this would be the best solution. Unfortunately in almost all African Countries, perceived sovereignty and not "bowing the knee" to the West is put at far higher premium than things like drug approvals. This would be scoffed at accross the continent for this reason.
To be fair it's not like high income country's are doing great at getting their approvals sorted, partly for similar pride reasons.
Yes, sadly I think you're right, and the fact that this would be a good policy for Western countries also probably makes little difference in the rhetorical calculus.
I however think it's important for African countries to have their own independent regulatory authorities because we've seen examples of first line drugs that work great in the West not necessarily work as efficiently for African populations, for example in antihypertensives, anti failure drugs etc. I thus believe African countries developing their own health data is key in the long run and a focus on streamlining market accessibility for new drugs THAT WORK FOR AFRICAN POPULATIONS is key.
Hi Larks, really appreciate your question. A few thoughts:
This directly addresses review delay but not submission delay. Outsourcing decisions to the FDA/EMA/etc would speed things up once an application is submitted by the manufacturer, but the manufacturer still needs to apply in the first place. The AMA tries to address both by making Africa more attractive as a unified continental market. You could argue that guaranteed automatic approval would give manufacturers every reason to submit (this feels intuitively right), but the EAC pilot undermines this somewhat, since Roche only registered its drugs in 3 of the 6 participating countries, even under a streamlined framework, so other market forces are clearly at play.
Drug approval is a context-specific risk-benefit judgment. The FDA approves drugs with American patients in mind, the EMA for Europeans. That decision may not translate to other contexts. This is why the EMA runs a separate program called EU-Medicines for All (EU-M4all) to assess medicines specifically for LMIC contexts. That said, it would be worth examining the actual data. It would be cool to have a list of drugs where approval decisions genuinely diverged based on population-specific factors, to get a sense of how often this is really a deciding issue in practice. I'll look into it!
Outsourcing approval doesn't outsource post-market surveillance. This matters especially in Sub-Saharan Africa, where substandard medicines are a serious problem.
The accountability gap is tricky. If a country gives up on doing their own drug approvals and solely relies on e.g., the FDA's decisions, it inherits the outcomes without much recourse. The FDA bears no responsibility for what happens in markets it didn't regulate for, so if something goes wrong, there's no clear mechanism for response.
One of the AMA’s long term goals is to strengthen national agencies by increasing their regulatory capacity. Outsourcing approvals could help with access in the near term, but without a complementary solution, African countries would indefinitely rely on others.
I think you can just skip the submission step. There's no need to require submissions at all. My submitting to the FDA or equivalent, you're automatically deemed to have submitted.
Yes, there are some context-specific elements, but they seem relatively small. Notably, once the FDA approves a drug, it is legal to use for any indication, and for any patient, in the US - even those the FDA did not label it for. I understand that there are biological differences in drug responses between different ethnic groups, but these are typically not that large, and note that the FDA already has a large population of African Americans, with ancestry from all across the continent, within its jurisdiction. Supply chain limitations in Africa, heat-resiliency, disease ecology etc. are real issues. But do they really justify multi-year delays for ordinary treatments? I think it is very unlikely that the cost-benefit analysis would come out this way. The EU runs EU-M4all, but this is focused on products that are focused on non-EMA markets - it doesn't mean than normal drugs approved for use in the EU are inappropriate for Africa.
To the extent that post-approval surveillance is a problem, it is actually good to outsource approval to developed countries, because this would free up scarce resources for surveillance.
The idea of an accountability seems more like a rhetorical / nationalist issue than a real one to me. The current degree of accountability national african regulators face seems pretty small in practice, and your preferred solution - an Africa-wide version - would also have accountability issues. I think ordinary Africans would be better served by a competent and (relatively!) swift international drug authority with no formal accountability to them over less-competent and slower national regulators with a veneer of accountability.
It really doesn't seem that bad to me for African countries to indefinitely rely on others? Specialization, Division of Labour and Gains from Trade are some of the most important drivers of a prosperous modern world. But even if drug-approval-capabilities was something that autarky was desirable for, these countries should build the competence first, rather than imposing onerous restrictions on their citizens - and causing many premature deaths through denying safe and effective drugs. I would rather have them build capacity by focusing on surveillance, inspections, trial oversight, and procurement integrity.
Executive summary: The author argues that fragmented and under-resourced drug regulation in Africa causes multi-year delays in access to essential medicines, and while regional coordination like the African Medicines Agency could reduce this, its success is uncertain and depends on political will, trust, and incentives.
Key points:
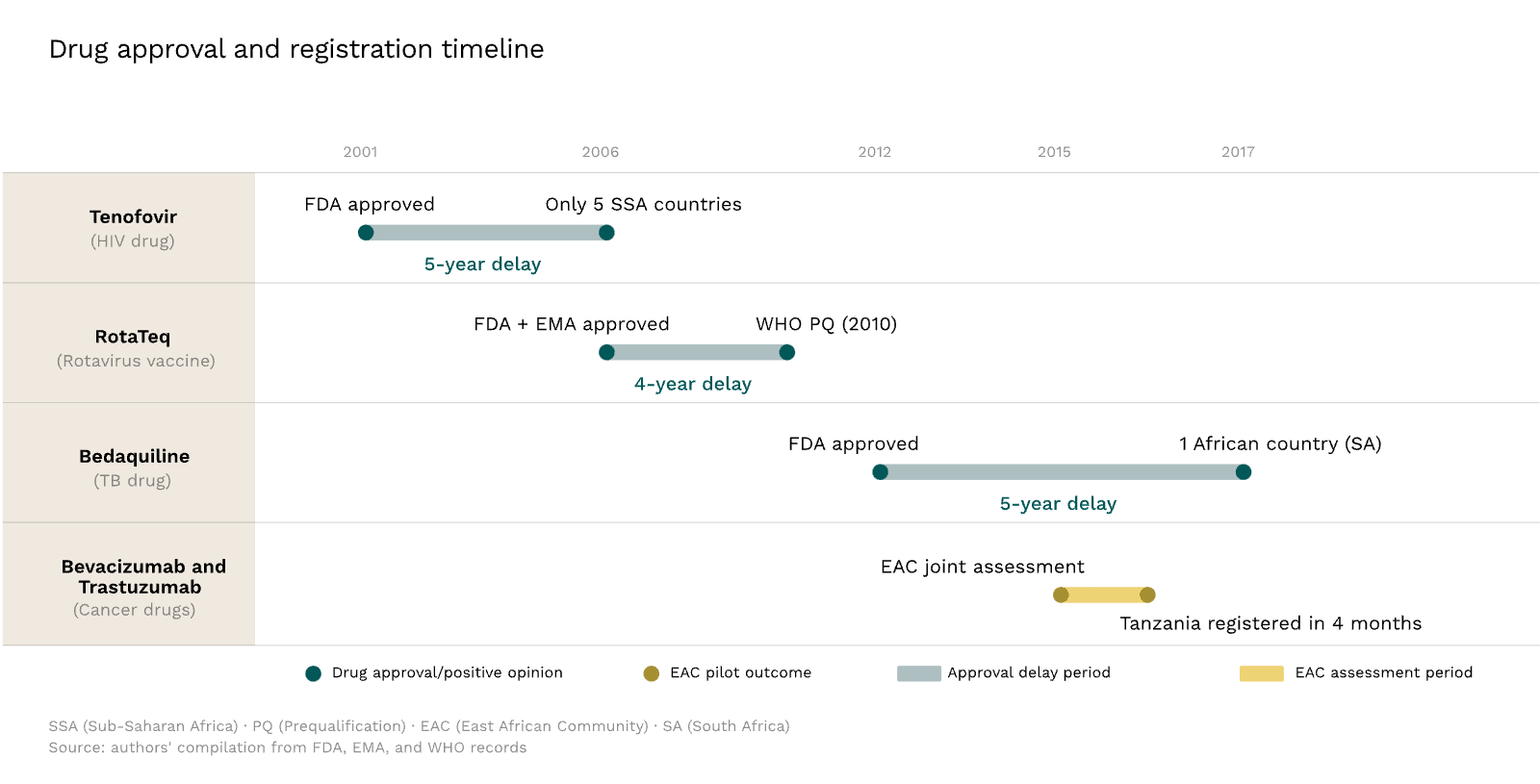
The author argues that regulatory delays in Africa—often 4–7 years after high-income approvals—have led to worse health outcomes, as seen with tenofovir and bedaquiline reaching patients years late despite clear benefits.
These delays are driven by submission barriers (firms must file separately in 55 countries with low financial incentive) and review barriers (limited regulatory capacity leading to multi-year approval timelines).
Cross-border “family style” regulation—harmonisation, collaborative review, reliance, and supranational models—can reduce duplication, but each has trade-offs, especially around trust and dependence on timely lead regulators.
The East African Community pilot showed that harmonisation and shared review can cut timelines (to ~240 days) and improve standards and capacity, but did not fully solve issues like selective market entry, slow national approvals, or lack of trust in joint decisions.
The African Medicines Agency (AMA) aims to scale this model continent-wide to improve coordination and health sovereignty, but remains a voluntary harmonisation body without binding authority, limiting its leverage.
The author argues the AMA’s impact will depend on whether it creates enough value for manufacturers and member states—given funding uncertainty, uneven participation (notably missing major markets), and risks of becoming an additional bureaucratic layer.
This comment was auto-generated by the EA Forum Team. Feel free to point out issues with this summary by replying to the comment, andcontact us if you have feedback.
This is a crosspost of the full text of Can Africa regulate as a continent?from In Development, made for the EA Forum's In Development Highlight Week.
If you enjoy the article, you can subscribe to In Development's substack here.
The problem of regulatory delay
The drug tenofovir disoproxil fumarate, a cornerstone of HIV treatment, was approved by the United States Food and Drug Administration in 2001. Its better safety profile quickly made it a standard treatment in the US and Europe. It should have been a shoo-in in Africa; at the end of 2001, sub-Saharan Africa accounted for over 70% of the world's HIV/AIDS cases, while an estimated 2.3 million people on the continent died of the disease that year.
Instead, when African countries began rolling out their national programmes to address the AIDS epidemic, with Botswana leading the way in 2002, most patients started on stavudine-based regimens.[1] Stavudine was cheap but toxic, causing disfiguring and sometimes life-threatening side effects. In South Africa, one study showed that 30% of patients stopped taking it within three years.
By early 2006, the manufacturer, Gilead, had registered it in only five sub-Saharan African countries. Even in South Africa, one of the region's larger markets, the company did not apply for registration until late 2005.
In Africa, programmes only began replacing stavudine with tenofovir in around 2010.[2] Had Gilead filed in African markets alongside the FDA, tenofovir could have been available from the start.[3]
This same pattern was repeated with bedaquiline, the first new class of tuberculosis drug in over 40 years. Following fast-track approvals by the FDA in 2012 and the European Medicines Agency in 2014, it was hailed as a breakthrough against drug-resistant tuberculosis, a disease that was killing over a million people annually. Most of those people lived in Africa and Southeast Asia. Yet, by October 2014, it had been registered in just one African country (South Africa). In South Africa, mortality among drug-resistant TB patients was roughly half on bedaquiline-based regimens compared to standard treatment. Elsewhere, patients continued to receive inferior drugs.[4]
The nature of regulatory delay
On average, in sub-Saharan Africa, there is a gap of four to seven years between a drug or vaccine's first submission to a regulatory agency in a high-income country and its approval. Two distinct regulatory barriers drive this.
The first is submission delay. Africa has 55 countries, and manufacturers must usually file separately in each one. This means repeated applications, different technical requirements, and higher costs. For firms weighing returns in any single market, the arithmetic often does not favour registration. They focus on larger, richer markets instead, so products are either not submitted or arrive years later. That was tenofovir's fate.
The second is review delay. For example, if you submit a drug for review in Botswana, realistically, you can't expect to start selling it until three years later. This is much longer than in countries like the US, where a standard review takes 10 months.
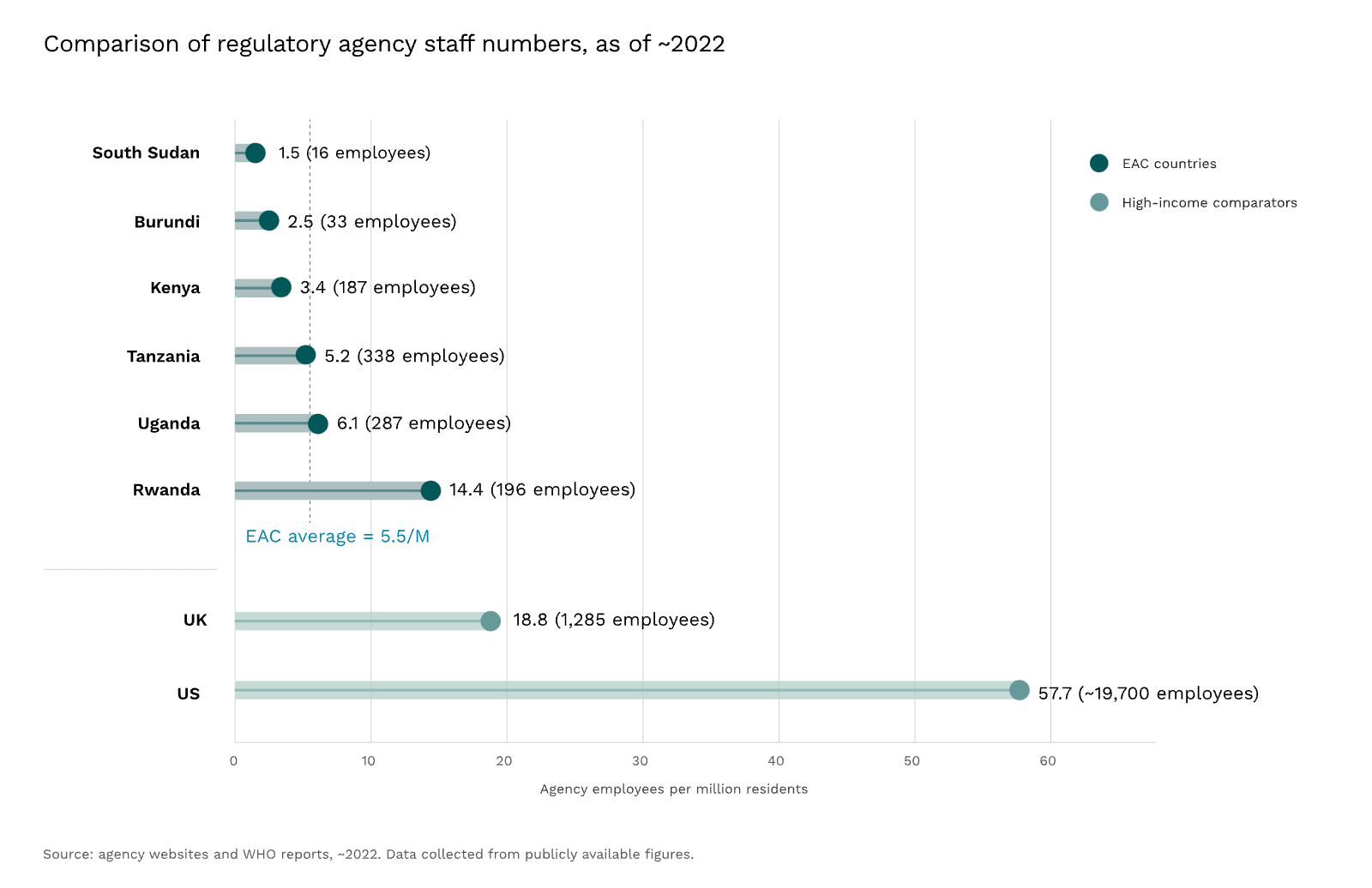
Drug reviews can be slow anywhere; the FDA, for instance, missed its own review deadlines for about 1 in 10 products in 2025. However, in developing countries, insufficient staffing at the regulatory agency can be a binding constraint. Reviewing a complex drug dossier requires trained pharmacologists, toxicologists, and clinical experts, as well as laboratory capacity to test product samples and systems to track adverse events once drugs reach patients. Most African countries lack this infrastructure.
According to the WHO, more than 90% of African countries have minimal to no regulatory capacity. For instance, in 2022, South Sudan—a country of about 11 million people—had just 16 staff at its medicines regulatory agency (~1.5 per million residents). By contrast, the US employed about 19,700 (~56 per million residents).
Regulating "family style"
Africa is not the only continent with many small countries, nor is it the only one that has faced limited capacity. Regulators around the world have developed three broad responses to submission and review delays. Each has involved some form of cross-border cooperation—call it regulating "family style."
Harmonisation of regulatory requirements across agencies addresses submission delays. When regulators standardise procedures, guidelines, and technical requirements, they make it easier for manufacturers to submit in multiple countries. The International Council for Harmonisation has spent decades aligning technical standards across major developed country markets, but much of Asia, Africa, and Latin America remains outside its framework.
Collaborative review goes further: regulators jointly assess applications while retaining national authority. The FDA's Project Orbis does this for cancer drugs. Since May 2019, the US, Australia, Canada, Singapore, Switzerland, the UK, and others have conducted concurrent reviews, often issuing approvals within days of each other. But this model depends on trust, which is easier when participating agencies have similar levels of capacity.[5]
Reliance reduces duplication by allowing regulators to defer to trusted authorities. For example, the UK can fast-track approval of drugs already authorised in other major markets.[6] The WHO Prequalification Programme operates on similar principles, allowing countries to rely on WHO's assessment rather than conducting their own full review.
However, reliance is only beneficial when the leading authority's reviews are timely. In 2022, WHO's full review pathways averaged about 17 months, a reminder that concentrating regulatory work in a single body amplifies the cost of that body's failures across reliant countries.[7]
The deepest form of collaboration is supranational regulation. In the European Union, the EMA conducts a single scientific review, and the European Commission issues one authorisation valid across all 27 member states. This only works because EU countries agreed to pool sovereignty for drug approval. Without that political foundation, the model is hard to replicate.
The East African pilot
In 2009, the African Union established a Medicines Regulatory Harmonisation initiative.[8] Rather than attempting continent-wide harmonisation and collaboration immediately, it decided to run a five-year pilot through one of the continent's existing regional economic communities, soliciting proposals and then funding the most promising plan. The East African Community (EAC) won the bid. Its application was compelling: it already had a customs union and a common market in force, giving its member states genuine experience in cross-border cooperation; it comprised a small number of partner states, most of which shared a common language, culture, and infrastructure; and its national regulators had already been collaborating informally for years.
And so, in 2012, the EAC's Medicines Regulatory Harmonisation (MRH) initiative was launched, covering nearly 150 million people. The initiative targeted reducing submission and review delays by adopting two of the "family style" regulatory approaches: harmonising requirements and speeding up review by sharing the work among national regulators, while maintaining rigorous standards.
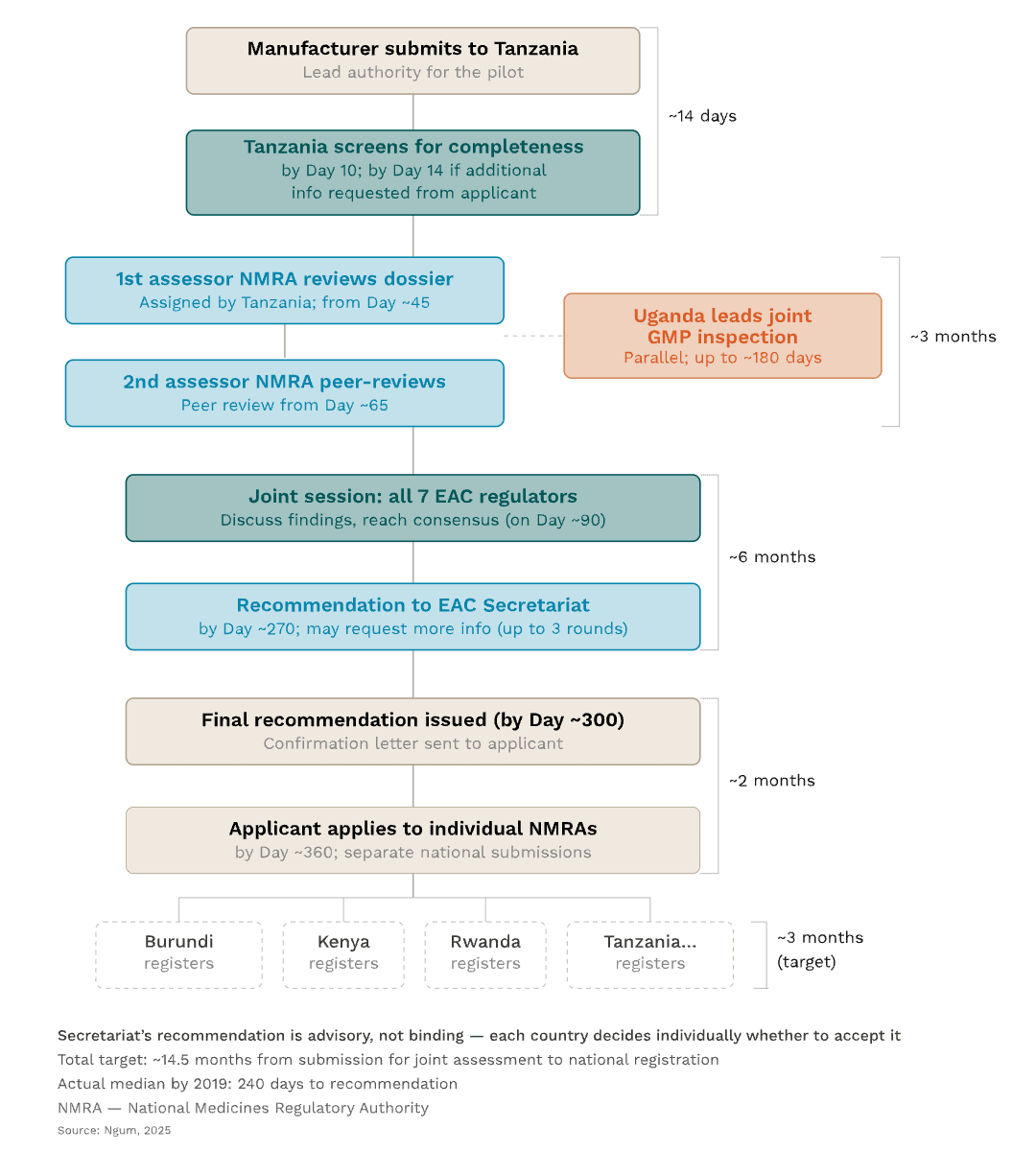
It was not designed as a supranational regulator issuing binding approvals. Applications would be jointly assessed, but final decisions on whether to approve a product would remain at the national level. There would be no central authority for medicine approvals in East Africa.
But it did split the work across countries. A product application was first submitted to Tanzania's regulatory agency, which was responsible for the initial screening to confirm that all sections were complete. Tanzania then assigned two other national authorities to evaluate the application's data in full, while Uganda's regulator simultaneously led the product's Good Manufacturing Practice assessment. Once these assessments were complete, all EAC regulators came together in a joint session to discuss the findings and reach a consensus recommendation. This was then submitted to the Secretariat, and the manufacturer could use it to apply for national marketing approval in each EAC member state individually.
And it worked. Up to a point.
The median timeline for joint assessment fell from about two years to just over a year by 2017, and down to 240 days by 2019. Between 2015 and 2020, the initiative held 10 joint assessment sessions, reviewing 83 product applications, and recommending 36 products for approval in the region. The initiative also succeeded in advancing regulatory harmonisation, by developing a Common Technical Document that manufacturers could use for submissions across all partner states. Joint Good Manufacturing Practice inspections began in 2016, pooling expertise and reducing redundant factory visits.
As well as reducing regulatory delay, the joint assessments introduced higher manufacturing standards than many national pathways required. They mandated bioequivalence studies, to demonstrate that generic drugs perform in the body in the same way as branded originals do. National procedures often waived such requirements, but the MRH had the capacity to insist on these.
Beyond speeding up review times, the initiative also sought to encourage drug classes that might not otherwise have been registered. Major global health organisations, such as the WHO, naturally prioritise advancing medical products to fight the highest burden diseases in Africa. These are usually infectious diseases. But as the African population ages, and the burden from infectious diseases has been reduced, non-communicable diseases have become increasingly important. The East African pilot decided to also focus on drug classes that treat these disease types, particularly anticancer and antihypertensive medicines. Before the pilot, such drugs were systematically under-registered: when Kenya's government attempted to procure essential cancer drugs, nearly a quarter weren't available in the country. Early joint assessment application data suggests the pilot began to address this gap: between 2015 and 2017, 16% of 49 applications were for oncology drugs and 24% for cardiovascular products.
Perhaps most importantly, the initiative helped build regulatory capacity across the region. When it began, only Kenya, Tanzania, and Uganda had regulatory agencies separate from their ministries of health; Burundi, Rwanda, and Zanzibar, by contrast, had small departments housed within theirs. By cooperating with more established regulators, Rwanda and Zanzibar were able to increase their independence.
But the limitations were equally apparent.
In 2015, Roche used the initiative to seek approval for two established cancer medicines, bevacizumab and trastuzumab.[9] The joint assessment proceeded quickly, with a positive recommendation issued within months of submission. Tanzania registered them within four months of the drugs' application for joint assessment, a remarkable improvement over its 15-month average.
Yet Roche registered the medicines in only three of the six countries under the initiative at the time. It chose not to pursue the smaller markets. Even with a streamlined process, manufacturers still faced separate national submissions and fees; these markets simply weren't worth it for Roche.
And the programme didn't fix all regulatory issues. National registration was supposed to take about three months, but sometimes took over a year. When pharmaceutical executives were surveyed about the initiative, their response was measured. They supported its ambitions and noted progress. But final national authorisations still took too long, and some countries failed to recognise joint recommendations.
This last point revealed a deeper tension. The premise of collaborative review is mutual trust. But some national regulators refused to accept the joint decisions. While their rationale isn't publicly known, it could have been due to a lack of trust. When sharing work across countries with very different capacity levels, those with high capacity may not wish to defer to those with lower capacity.
The introduction of higher standards for generics created its own pressures. During the pilot, the industry became frustrated that the joint regional standards were higher than those previously applied in some member countries. As long as some countries maintain less demanding requirements, regulatory arbitrage—where firms capitalise on regulatory loopholes to avoid unfavourable rules and cut compliance costs—becomes possible, and companies may simply choose to submit only to countries with lower standards.
And there was one other issue. The initiative was meant to become self-sufficient after five years, transitioning from donor funding to fees and contributions from partner states. Nine years later, that transition hasn't happened. Relying on outside funding introduced more delays to the process, and made the whole endeavour fragile.
Scaling to a continent
Still, the East African pilot was always intended to lead to something larger. Conversations about a continental regulator date back to 2009, but it took a decade of political negotiation before the African Union formally adopted a treaty establishing the African Medicines Agency (AMA). Scaling the harmonisation model to 55 countries had the potential for the same gains seen in East Africa, but it also meant confronting the same obstacles, compounded across a far larger and more diverse region.
Even after treaty adoption, creating the AMA wasn't easy. In 2020, not long after the agency was created, the continent faced the COVID-19 pandemic. Most African countries lacked sufficient regulatory capacity to handle the approval of new medications and therapies. Instead, they had to depend on authorisations from the FDA, EMA, and WHO, leaving them with little autonomy over which vaccines and treatments they could access—or when. For proponents of the AMA, it was a concrete illustration of what a continental regulator was meant to address. Well-resourced regulators can supplement domestic capacity, but they cannot substitute for it. The AMA is intended as a coordination mechanism designed and governed by African states themselves, rather than relying indefinitely on external authorities. In that sense, the AMA fits within a broader African Union commitment to improve medical capacity on the continent and achieve health sovereignty.[10]
By the time the AMA officially launched in November 2025, some 39 member states had signed or ratified the treaty. But 16 countries, including South Africa and Nigeria, two of the continent's largest pharmaceutical markets, had yet to commit, preferring to maintain independent regulatory frameworks. Without them, the AMA is likely to face considerable headwinds, operating with a meaningfully smaller share of total African pharmaceutical trade and a much weaker incentive for companies to engage with the agency at all.
The AMA also faces another significant hurdle. It is a harmonisation initiative; it is not a supranational regulator—at least for now. Participation is voluntary, and countries retain the right to disregard its assessments entirely. Despite frequent comparisons, it is not "an EMA for Africa." If engaging with the AMA and the individual countries costs more time and money than simply submitting to a country directly, companies will take the simpler path. If that isn't the AMA, the agency risks becoming regulatory theatre, significant on paper but inconsequential in practice.
The AMA can draw a clear lesson here from the East African pilot. An industry survey found that most manufacturers had expected joint assessment decisions to be automatically accepted by individual national regulatory authorities. Manufacturers who had expected automatic acceptance lost confidence in the programme when they realised that this was not what it did. Setting expectations at the outset and clearly communicating the actual benefits of the AMA will position the continental agency far better with manufacturers than the pilot did.
As with the pilot, financing will also be a pressing question. Since 2022, the AMA has attracted significant external financial support—100 million euros over five years from the EU and the Gates Foundation, alongside contributions from Wellcome, the European Commission, and Belgium. But donor funding has expiration dates. The agency will ultimately require either substantial contributions from member states or it must begin to charge drug manufacturers.[11]
In many ways, the AMA will take all the challenges experienced in the pilot and amp them up. Building trust was difficult enough across countries in East Africa; it will be all the harder across the entire continent. Even the question of language becomes a serious operational problem: the EAC operates across three languages while the African Union officially recognises six.[12]
If it goes well, the AMA could become a trusted coordinator that reduces duplication and accelerates access across the continent. Or, if these tensions aren't resolved, it could become just another layer of bureaucracy, adding assessments, fees, and complexity without shortening national approval timelines.
Which outcome prevails will depend on whether the AMA can deliver value that justifies the additional step, and whether enough member states have the political will to let it try.
A worthy experiment
In many ways, the AMA is more than an experiment in drug regulation. It is an experiment in regional cooperation in the developing world—an experiment in which the stakes are high, resources are scarce, and incentives push toward yet more fragmentation.
The AMA is attempting something genuinely ambitious: regulatory coordination across 1.5 billion people, 55 member states, six languages, and enormous variation in capacity and political will. The EMA took decades to reach its current form, even with the advantage of operating within high-income Europe. Africa has neither time nor the advantages of money. The work will be technical, administrative, and often dull (to all but the most passionate regulatory wonks). But the alternative to a project like the AMA is that essential HIV treatments arrive half a decade late in places that needed them most.
Enlli McAleese is a researcher and advisor focused on strengthening medicines regulatory systems in Africa. She previously served as Regulatory Director at 1Day Sooner and worked on the COVID-19 vaccine rollout at the UK Department of Health & Social Care.
Before the adoption of tenofovir in Africa, first-line antiretroviral therapy primarily relied on older nucleoside reverse transcriptase inhibitors combined with either a non-nucleoside reverse transcriptase inhibitor or a protease inhibitor. Common first-line regimens were stavudine, lamivudine, plus either nevirapine or efavirenz.
It bears noting that registration alone may not have guaranteed access, since tenofovir's price remained prohibitive until Gilead's voluntary licensing programme in 2006 enabled generic production and dramatically reduced costs. Earlier registration, however, would have created the legal precondition for faster licensing negotiations and generic entry.
Vaccines follow a slightly different path. Typically, a vaccine needs to obtain WHO prequalification before Gavi, the Vaccine Alliance, will fund it and the United Nations Children's Fund (UNICEF) will procure it, meaning that delays at the WHO stage ripple forward and block access at scale. National registration adds a further layer, with countries in Sub-Saharan Africa taking an average of one to two years to register a vaccine even after WHO prequalification has been granted. Because Gavi's funding decisions are tied to WHO prequalification, the WHO prequalification delays tend to have an outsized effect on access compared to any individual country's regulatory timeline. For example, RotaTeq, Merck's rotavirus vaccine, was approved by the FDA and the EMA in 2006 but didn't receive WHO prequalification until 2010, leaving a four-year gap before Gavi could procure it for a disease that kills the vast majority of its victims in low-income settings.
Because of this dynamic, collaborative review has historically been used most often by countries with well-established regulatory agencies (such as countries that have WHO-Listed Authorities).
If Australia, Canada, the EU, Japan, Singapore, Switzerland, or the US has already licensed a drug, the UK's Medicines & Healthcare products Regulatory Agency, through its International Recognition Procedure, can issue local authorisation far quicker.
Ironically, this was largely due to limited resources—the same structural weakness that drives countries to outsource their reviews in the first place.
Similar efforts to strengthen regional medicines regulation are underway in other parts of the Global South. In Latin America and the Caribbean, the Pan American Network for Drug Regulatory Harmonization (PANDRH) is hosted by the Pan American Health Organization, and has facilitated regulatory harmonisation and reliance since 1999, and, in 2023, the Latin American and Caribbean Medicines and Medical Devices Regulatory Agency (AMLAC) was established. In Southeast Asia, a continental agency has been discussed within the Association of Southeast Asian Nations (ASEAN). Notably, PANDRH, AMLAC and the existing ASEAN regulatory network operate as looser networks centred on harmonisation and regulatory reliance rather than on centralised decision-making, and do not carry the same legal authority that the AMA derives from its founding treaty.
This was not the first time a health crisis had spurred the creation of an African institution. Before the 2014–2016 Ebola outbreak, proposals for a continental public health body in Africa had circulated for years. African leaders had formally acknowledged the need in 2013, but with little urgency. The scale of the outbreak changed that. The African Union's dependence on outside responders made the institutional gap impossible to ignore, and the Africa Centres for Disease Control and Prevention was established in 2017.
BLUF:
* To determine whether AI is ‘improving exponentially’, ‘hitting the wall’, or any other claim which involves a quantity or magnitude (e.g. ‘This model was a big leap/small increment’). We need a good y-axis: an interval scale of AI capability which means +1 unit always represents the same degree of ‘how much better’, in the same way +1 degree Celsius is always the same amount of ‘how much hotter’.
* Yet there is no good y-axis for AI capability. All our...
Public service announcement
1. Applications are now open for our first ever round of the Charity Entrepreneurship Incubation Program dedicated exclusively to animal welfare. Learn more about what’s different this round here and apply...
Summary
* The animal welfare movement has already seen an influx in funding and should prepare for the possibility of more.
* The EA Animal Welfare Fund is encouraging those working in animal advocacy to actively set aside time and resources now to concretely plan for scaling sustainably, and we’ll support you in doing that.
* We’re requesting advocates set concrete ambitious goals and submit plans t...

Thanks for sharing this, it was a very interesting read!
I do want to question this claim though:
It seems to me that an attractive alternative would be for African countries to simply give up on doing their own drug approvals, and outsource the decision making entirely to the FDA, MHRA, PMDA, EMA etc. Why not simply say that any drug approved by any of these agencies is automatically approved? This way drugs would be approved swiftly and with almost zero cost to both government and corporation.
100% agree this would be the best solution. Unfortunately in almost all African Countries, perceived sovereignty and not "bowing the knee" to the West is put at far higher premium than things like drug approvals. This would be scoffed at accross the continent for this reason.
To be fair it's not like high income country's are doing great at getting their approvals sorted, partly for similar pride reasons.
Yes, sadly I think you're right, and the fact that this would be a good policy for Western countries also probably makes little difference in the rhetorical calculus.
Hi Nick,
I do get the "perceived sovereignty" angle.
I however think it's important for African countries to have their own independent regulatory authorities because we've seen examples of first line drugs that work great in the West not necessarily work as efficiently for African populations, for example in antihypertensives, anti failure drugs etc. I thus believe African countries developing their own health data is key in the long run and a focus on streamlining market accessibility for new drugs THAT WORK FOR AFRICAN POPULATIONS is key.
Hi Larks, really appreciate your question. A few thoughts:
Thanks for replying!
Hi!
I didn't know about the EU medicine for all and I think that's really cool.
I enjoyed reading through the article